A few (minor) complications
DNA consists of an inter-leaved helix of two strands
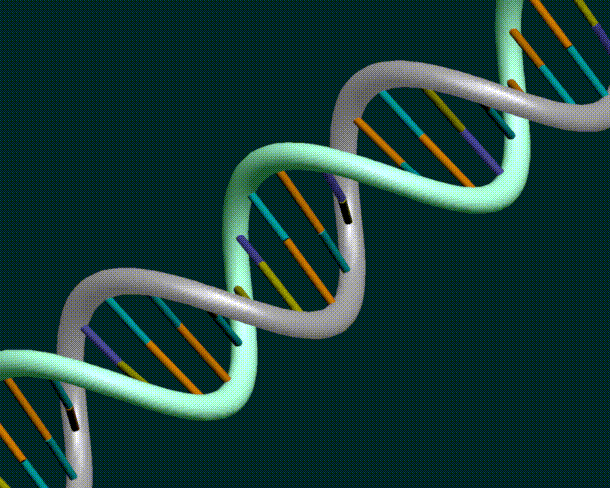
DNA sequences from the NBCI database only have one strand of the DNA. However, given one strand, it is a simple matter to derive the other strand.
DNA searching
Also, it’s unclear which direction the sequence was captured. So…
• There are four strands to consider when searching the genome.
•1. The original strand from the database. E.g. Strand 1, forward direction.
•2. Strand 1, reverse direction. (flip #1 )
•3. Strand 2, forward direction (Derive this from #1 above) via simple rules.
•4. Strand 2, reverse direction (flip #3 above.)
Optimization: Instead of creating and storing all 4 strands, it is possible to just adjust the query, e.g. a search for “CAT” could also result in these three other searches:
•TAC (reversed), GTA (second strand), ATG (second strand reversed)
•With this optimization, less memory is required to store the DNA information.