![]()
![]()
![]()
Charge-transfer photochemistry via electron donor/acceptor organizations
Jay K. Kochi, Department of Chemistry, University of Houston, 4800 Calhoun Rd., Fleming Building, Houston, TX 77204-5641, Fax: 713-743-2709, jkochi@pop.uh.edu
Photoexcitation of various intermolecular electron donor/acceptor complexes via the specific irradiation of their charge-transfer absorption bands leads to a wide variety of transient ion-radical pairs (CIRP) in their ground states.
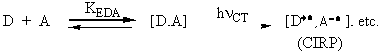
The subsequent fate of the CIRP determines the course of charge-transfer
photochemistry. Such CIRPs are highly reminiscent of contact ion
radical pairs formed in the corresponding thermal (adiabatic)
electron transfer between D and A especially via the inner-sphere
mechanism. As such, the direct bearing of charge-transfer activation
to the transition states of many common organic reactions will
be described.
Supramolecular and magnetic control effects in organic photochemistry
Nicholas J. Turro, Department of Chemistry, Columbia University, 3000 Broadway, Mail Code 3119, New York, NY 10027, Fax: 212-932-1289, turro@chem.columbia.edu
During the last two decades the complexity of organic photoreactions has been manifest in the use of supramolecular and magnetic effects to control the course of photochemical reactions involving radical pairs. Examples will be given of (1) the use of EPR to directly detect and characterize geminate radical pairs in supramolecular systems, (2) the use of supramolecular systems to control the reaction pathways of geminate radical pairs, and (3) magnetic isotope control of the reaction pathways of radical pairs.
Nanocrystalline titanium dioxide solar cells sensitized with ruthenium or osmium polypyridyl complexes: Photoelectrochemical studies and electron transfer dynamics
Darius Kuciauskas(1), Genevieve Sauvé(1), Michael S. Freund(2), Harry B. Gray(2), Jay R. Winkler(2), and Nathan S. Lewis(1). (1) Division of Chemistry and Chemical Engineering, California Institute of Technology, 1200 E. California Blvd. m/c 127-72, Pasadena, CA 91125, darius@caltech.edu, nslewis@caltech.edu, (2) Beckman Institute, California Institute of Technology.
Novel nanocrystalline semiconductor solar cells sensitized with transition metal polypyridyl complexes achieve efficiencies of up to 10% and are promising for applications in photovoltaics. These systems are also important for fundamental photochemical studies of interfacial electron transfer between molecular sensitizers and wide band gap nanocrystalline semiconductors.
A series of osmium polypyridyl complexes having various ground-state reduction potentials has been synthesized and used to sensitize nanoporous titanium dioxide electrodes to solar illumination. Exposure of TiO2 electrodes to sources of OsII(H2L')2(CN)2, (where L' is 4,4'-dicarboxylato-2,2'-bipyridine) or OsII(H2L')32+ extended the light absorption and spectral response of the cell to longer wavelengths than did exposure of TiO2 to Ru(H2L')2(NCS)2. The Os complexes also provided very high external quantum yields for photocurrent flow and produced open-circuit voltages similar to those of the Ru complex.
The electron transfer dynamics in these systems have been studied on the femtosecond to millisecond time scales. The ruthenium and osmium bipyridyl complexes Ru(H2L')2(CN)2, Os(H2L')2(CN)2, Ru(H2L')2(NCS)2, and Os(H2L')2(NCS)2 inject electrons into the semiconductor with the rate constants 1013 - 1010 s-1. Electron injection was observed from both singlet and triplet MLCT states, and the electron injection rate is sensitive to the spin-orbit coupling in the sensitizer complex. Charge recombination reactions are much slower, which ensures high efficiency of nanocrystalline solar cells. The effects of excitation intensity, temperature, and applied potential on the recombination reaction were analyzed using a second-order kinetics model. The rates of charge recombination decrease with increasing driving force to the oxidized sensitizer, indicating that charge recombination occurs in the Marcus inverted region. The electronic coupling factors between the oxidized sensitizer and the injected electrons in the TiO2 and the reorganization energies for the recombination reaction vary significantly for the different metal complexes. The charge recombination rates are well-described by semiclassical electron transfer theory, and reorganization energies are 0.55 - 1.18 eV. The observation that charge recombination occurs in the Marcus inverted region has important implications for the design of molecular sensitizers in nanocrystalline solar cells operated under our experimental conditions.
Photoprocesses in ordered thin films
Thomas L. Penner, Imaging Materials and Media, Research and Development, Eastman Kodak Company, 1669 Lake Avenue, Rochester, NY 14652-4708, thomas.penner@kodak.com
The photochemical and photophysical requirements of materials for optical and photonic applications can be very demanding and specific. Although fundamental molecular properties are essential to achieving desired performance, intermolecular interactions and larger scale organization can be used to enhance existing properties and even generate new useful ones. To obtain or to engineer such effects requires that the building-block molecules have other functionality in addition to interacting with light in a specified way. Because the resulting systems usually consist of deliberately interactive multiple components, there is both the opportunity and the challenge of optimizing conditions to maximize performance. These issues and advantages to be gained from such organized interactive materials will be illustrated with examples that demonstrate that "photochemistry becomes more complex" for current technological applications. In keeping with a symposium honoring George Hammond, photoactive organic materials will be central to this presentation.
Extracting fundamental (simple) photochemical and photophysical information from reactions of guest molecules in complex polymeric media
Weiqiang Gu, and Richard G. Weiss, Department of Chemistry, Georgetown University, Washington, DC 20057-1227, Fax: 202-687-6209
Complex polymeric media offer unique opportunities to obtain subtle mechanistic information from (as well as to direct the courses of) photophysical processes and photochemical reactions of guest molecules. However, extracting such information and understanding why selective pathways are followed present formidable challenges. Analyses of results from the photo-Fries reactions of some aryl esters and photo-Claisen reactions of some aryl ethers in polyolefinic films and 'model' isotropic media will be presented as examples. Factors associated with the directing influences of the reaction 'cages' of the polymers on singlet radical-pair intermediates in these reactions will be discussed. They include (1) small differences between the trajectory of approach of phenylacyl and benzylic radicals when bonding to their aryloxy radical partners in a cage and (2) the intrinsic reactivity of the aryloxy radicals. Our inability to understand fully the interplay between the polymeric hosts and their guests begs the question, "Has photochemistry become more complex or have photochemists become simpler?"
Probing enzymes with photosensitizers linked to substrates
Harry B. Gray, Alex R. Dunn, Ivan J. Dmochowski, and Brian R. Crane, Beckman Institute, California Institute of Technology, 1200 E. California Boulevard, Mail Code 139-74, Pasadena, CA 91125-7400, Fax: 626-449-4159, hgcm@its.caltech.edu
Cytochrome P450cam (P450) substrates tethered to luminescent Ru sensitizers bind the enzyme with affinities comparable to or greater than those of the unmodified substrate. The luminescence lifetime in the Ru-linked substrate is a critical indicator of enzyme binding; Ru-Fe(heme) energy transfer in the bound complex dramatically accelerates excited-state decay. Dissociation constants for P450:Ru-substrate conjugates have been determined from analyses of luminescence decay kinetics. The Ru-Fe distance extracted from the energy-transfer kinetics in one conjugate is in excellent agreement with that found in a single crystal. The similar Ru-Fe distances calculated for various Ru-substrates suggests that substrates bind via a common access channel. Luminescence-decay measurements indicate that these Ru-substrates bind selectively to P450 in the presence of several other heme proteins. Sensitizer-linked substrates could form the basis for a new class of optically detected in situ biosensors.
Photochemical industry becomes complex, too
David F. Eaton, DuPont iTechnologies, DuPont Experimental Station, Bldg 334, PO Box 80334, Wilmington, DE 19880-0334, David.F.Eaton@usa.dupont.com
In the beginning, photochemistry was simple, and so were its applications in industry. Photopolymer processes to produce polymeric printing plates, methods to preview the quality of printed color artwork (proofing), and ways to produce printed wiring boards (PWB's) and to build semiconductor integrated circuits using high resolution photoresists were invented and commercialized during the 1960's and 70's. These inventions revolutionized the way printing and publishing was accomplished by eliminating hot lead type and laborious and costly press proofing. PWB's and semiconductor resists enabled the electronics industry as we know it today.
And then things became complex for industry, just as they were becoming complex for photochemistry in general.
New applications of photochemical technology will be commercialized in the next years. These technologies will be complex and difficult to implement, but they will revolutionize the way we live, work and play just as the earlier inventions in a simpler time have made life today what it is now. This talk will provide a personal view of where "applied photochemistry" is going, and why it is a good thing that the world becomes more complex.
Photoreactions studied by step-scan transient IR spectroscopy
Douglas C. Neckers(1), M. A. J. Rodgers(2), Andrei Federov(1), and Eugene Danilov(1). (1) Center for Photochemical Sciences, Bowling Green State University, Bowling Green, OH 43402, Fax: 419-372-0366, neckers@photo.bgsu.edu, (2) Center for Photochemical Sciences/Department of Chemistry, Bowling Green State University
Step-scan transient IR spectroscopy provides infrared spectra from a photochemical process within approximately 20 ns following flash excitation. This paper will report work in the author's labs that uses TR-IR to relate structure to the creation and destruction of reaction intermediates. Several examples will be reported and discussed.
Laser photochemistry: 25 years toward a fulfilled promise?
James T. Yardley, Columbia Radiation Laboratory, Columbia University, 1020 Schapiro Center (CEPSR), Department of Chemical Engineering and Applied Chemistry, New York, NY 10027, Fax: 212-854-1909, jy307@columbia.edu
Twenty-five years ago, the chemical world was enthralled by the discovery of the laser - a source of an infinite quantity of cheap photons perfectly defined at any desired wavelength. The initial euphoria for laser chemistry quickly faced a number of very basic problems, involving both technology and economics. When the scientific world began to recognize some of the economic realities of laser photochemistry, it turned toward "high value chemistry": isotope separation, pharmaceuticals, and catalysis. Even for these applications the opportunities of laser photochemistry did not prove to be commercially viable. Some creative and forward-looking scientists recognized the possibilities for application of laser photochemistry in the realm of semiconductor processing. I will describe some of this early activity including the development of new photoresist materials and photochemical processes directed specifically toward laser processing. Although it may be less obvious at first glance, the creation and development of guided wave optical devices provides another area of major opportunity for laser photochemistry. Much of this work has derived from the leadership and from the philosophical and scientific directions brought by George Hammond to AlliedSignal, Inc. I will review work in this realm carried out over the past 10 years.
Photochemistry offers the opportunity to directly define the spatial variation of refractive index within a polymeric medium. This ability to precisely define refractive index profile provides the ability to create devices that correspondingly direct and control the propagation of light within the medium. Looking into the future it is clear today that laser photochemistry will provide the key for development of the next several generations of semiconductor devices through short wavelength laser lithography. Laser photochemistry will also be seminal for the fabrication of low cost optical devices for telecommunications as well as for a host of data communication applications. I will explore these opportunities and I will speculate on other possibilities. These observations strongly support the hypothesis that laser photochemistry has fulfilled its initial promise - perhaps not so quickly as initially envisioned and perhaps in ways not widely appreciated 25 years ago.
Leitmotif from the life of a photochemist
Eve L. Menger-Hammond, Department of Chemistry, Portland State University, P.O. Box 751, Portland, OR 97207-0751, Fax: 503-725-3888, evemenger@home.com
George S. Hammond's career has spanned nearly 50 years and touched several institutions and many lives. This retrospective highlights that career and examines his creativity in science, philosophy, and elsewhere.
![]()